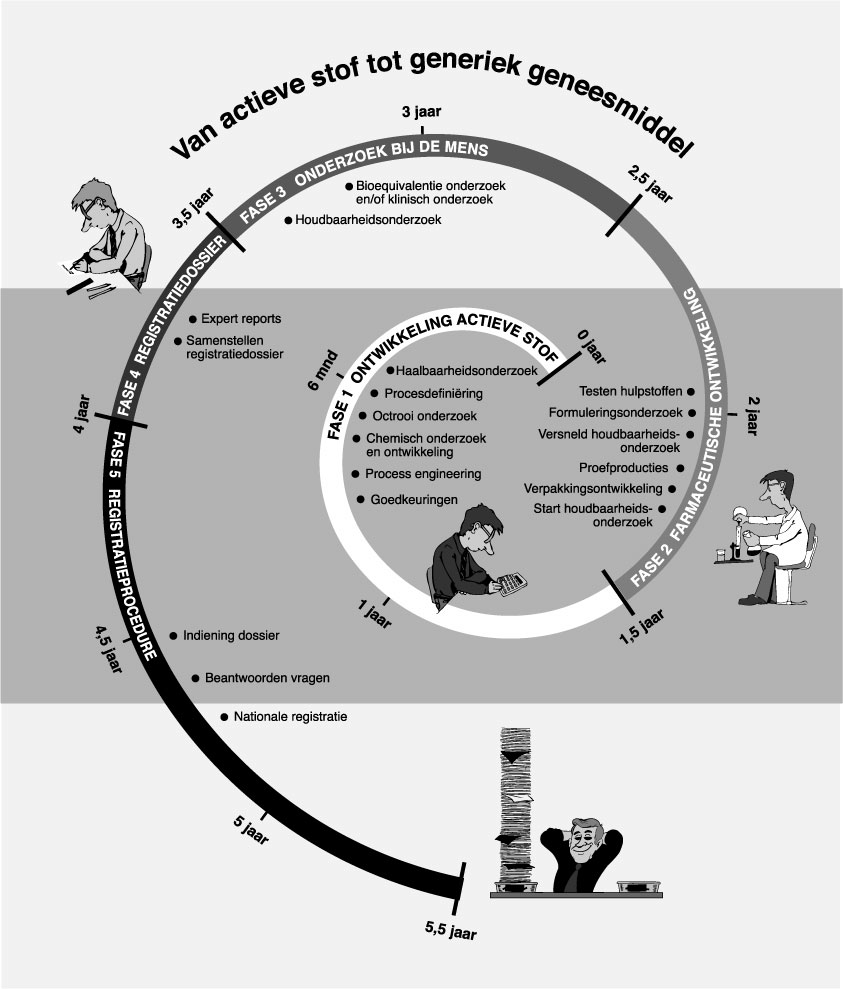
Hoe wordt het generiek geneesmiddel ontwikkeld?
Inleiding
De ontwikkeling van een actieve stof tot generiek geneesmiddel vereist veel tijd. Gemiddeld duurt het 5,5 jaar voordat het geneesmiddel op de markt komt (inmiddels is dit gemiddelde gedaald tot circa 4 jaar). Een generiek geneesmiddel is gemaakt van een werkzame stof, waarvan het octrooi op de synthese verlopen is. De ontwikkeling is nodig om een gelijkwaardig geneesmiddel te kunnen samenstellen.
Gemiddeld wordt door generieke geneesmiddelenfabrikanten 7,4% van de omzet besteed aan Research
& Development (Bron: Global Generics Guide, Datamonitor).
De wetgeving binnen de Europese Unie laat het niet toe vóór octrooiafloop van een specialité met dit ontwikkelingsproces te beginnen. Dit gebeurt daarom vaak buiten de Europese Unie, hetgeen nadelig is voor de werkgelegenheid en voor de concurrentiepositie van de generieke geneesmiddelenindustrie. De registratieperiode alleen duurt gemiddeld 1,5 jaar. Tijd, die door wetgeving ingekort kan worden waardoor het (voordeliger) generieke geneesmiddel sneller beschikbaar komt.
Hieronder worden de diverse fasen beschreven die leiden tot de productie van het generieke geneesmiddel.
Fase 1: Ontwikkeling actieve stof
Allereerst moet vastgesteld worden of het ontwikkelen en produceren van een bepaalde actieve stof commercieel aantrekkelijk en technisch haalbaar is. Daarnaast worden alternatieve syntheseroutes en de verkrijgbaarheid van grondstoffen voor de synthese bekeken. Hierna volgt het definiëren van het syntheseproces op kleine laboratoriumschaal. Tegelijkertijd wordt uitgezocht in hoeverre de voorgestelde synthese al dan niet inbreuk maakt op bestaande octrooien.
De volgende stap is dat op laboratoriumschaal grotere hoeveelheden (kilogrammen) worden gesynthetiseerd, waarbij de kritische procesparameters worden vastgesteld. Dan worden ook de analysemethoden ontwikkeld om de gesynthetiseerde verbinding te kunnen onderzoeken op zuiverheid en houdbaarheid. Proefpartijen worden op fabrieksschaal gesynthetiseerd waarbij de procesopschaling wordt gevalideerd. Valideren is vaststellen dat de methode voldoende gevoelig is en door verschillende instanties goed bruikbaar is bij herhaling.
Aan het einde van fase 1 stelt men alle benodigde kwaliteits-, proces- en vergunningsdocumenten op. Zo’n document heet bijvoorbeeld Drug Master File (DMF) in het geval van een andere synthese dan waarop bijvoorbeeld de Europese Pharmacopée (EP)gebaseerd is. Is de synthese route gelijk aan die van de Europese Pharmacopée , dan moet bij de verantwoordelijke autoriteiten een "certificate of suitability" worden aangevraagd.
Fase 2: Farmaceutische ontwikkeling
Toedieningsvorm
Als de actieve stof beschikbaar is gekomen (fase 1), kan worden gestart met de ontwikkeling van de toedieningsvorm. Mogelijke hulpstoffen worden fysisch-chemisch onderzocht waarbij bekeken wordt welke hulpstoffen te gebruiken zijn in combinatie met de actieve stof. Ook worden de analysemethoden die nodig zijn voor de keuring van de hulpstoffen en het gerede product ontwikkeld en gevalideerd en specificaties worden opgesteld.
Houdbaarheid
Nadat de hulpstoffen zijn gekozen, wordt de eerste hoeveelheid geneesmiddelen - d.w.z. de actieve stof in de beoogde toedieningsvorm - gemaakt. Deze eerste kleine hoeveelheid tabletten, capsules, crème of stroop etc. wordt onderworpen aan een zogenaamd versneld houdbaarheidsonderzoek (het zgn. ‘stresstesten’). Hierbij wordt het nieuwe product gedurende een aantal weken bewaard bij verhoogde temperaturen en vochtigheden, waarna de eventueel opgetreden ontleding wordt bepaald. De mate van ontleding geeft een voorspelling hoe het middel zich zal houden onder normale omstandigheden en wat de verwachte houdbaarheid zal zijn.
Proefpartijen
Als de uitslagen van dit onderzoek bevredigend zijn, worden de eerste proefpartijen gemaakt. Deze charges worden gemaakt met apparatuur die vergelijkbaar is met de machines die later gebruikt zullen worden bij de productie op grote schaal. De proefpartijen worden na de beginkeuring verpakt in verpakkingsmateriaal dat volledig vergelijkbaar is met het materiaal dat later gebruikt gaat worden als het product verkocht wordt. Hierna worden de proefpartijen opgeslagen voor het langlopende houdbaarheidsonderzoek. Op gezette tijden na het opslaan (bijvoorbeeld na 3, 6, 9 of 12 maanden) worden monsters van de partijen getrokken en gekeurd op hun kwaliteit.
Fase 3: Onderzoek bij de mens
Dit is de fase waarin bewezen moet worden dat de gekozen formulering equivalent (gelijkwaardig) is wat werking betreft aan het referentieproduct.
Voor orale geneesmiddelen als tabletten, capsules en dergelijke betekent dit een onderzoek naar de biologische beschikbaarheid. Hierin wordt bekeken of de hoeveelheid en de snelheid van opname van de actieve stof uit de toedieningsvorm even groot zijn als die van dezelfde stof vanuit het referentiepreparaat. Hiertoe worden de preparaten aan een aantal gezonde vrijwilligers gegeven en wordt de hoeveelheid van het geneesmiddel in het bloed en de urine over langere tijd bepaald. Er wordt vanuit gegaan dat een geneesmiddel dat gelijkwaardig biologisch beschikbaar is, ook gelijkwaardig werkt. Deze onderzoeken vinden plaats bij daartoe gespecialiseerde onafhankelijke instituten.
Uiteraard gelden hierbij de regels van Good Clinical Practice (GCP). Als de uitkomsten - statistisch betrouwbaar - vergelijkbaar zijn, spreken we van bio-equivalentie en zijn de preparaten onderling uitwisselbaar. In sommige gevallen is een dergelijk bio-equivalentieonderzoek niet mogelijk. Bijvoorbeeld als het geneesmiddel lokaal werkzaam is (huid, longen, etc.) en er geen bloedspiegels of urinewaarden te bepalen zijn. Dan worden er klinische studies bij patiënten uitgevoerd en de werking wordt vergeleken, uiteraard opnieuw door een onafhankelijk instituut en onder de regels van GCP. Equivalentie betekent in deze gevallen dat de preparaten dezelfde therapeutische effecten hebben, bijvoorbeeld een zelfde verbetering van de longfunctie bij inhalatiepreparaten. In de tussentijd loopt het houdbaarheidsonderzoek door en worden op gezette tijden monsters uit de partijen genomen en analytisch onderzocht. Verlopen alle onderzoeken naar wens, dan wordt in de volgende fase het registratiedossier voorbereid.
Fase 4: Het registratiedossier
Als de uitslagen van het bioequivalentie-onderzoek bevredigend zijn en ook het doorlopende houdbaarheidsonderzoek aantoont dat de gekozen formulering de juiste is, kan begonnen worden met het samenstellen van het registratiedossier. De indeling en de inhoud van dit dossier moeten voldoen aan alle eisen die in vele gedetailleerde Europese regelingen zijn neergelegd. Het registratiedossier is daardoor ook geschikt om te gebruiken om een registratie te verkrijgen in andere landen van de Europese Unie.
Het registratiedossier voor een geneesmiddel bestaat standaard uit vijf delen. Voor de registratie van een generiek geneesmiddel zijn met name van belang:
Deel I - samenvatting dossier, waarin de bijsluiter en wetenschappelijke tekst die moeten voldoen aan de eisen die het College ter Beoordeling van Geneesmiddelen (CBG) stelt aan de betreffende actieve stof (indicaties, contra-indicaties bijwerkingen en dergelijke).
Voorts bevat deel I de 'expert-reports'. In deze rapporten worden de resultaten van de onderzoeken in het kort van deskundig commentaar voorzien en worden keuzes die het bedrijf tijdens de ontwikkeling gemaakt heeft, toegelicht. Voor het dossier van een generiek geneesmiddel is vooral het ‘expert-report’ dat de onderzoeken en resultaten van deel II van een toelichting voorziet van groot belang. Daarnaast worden er ‘expert-reports’ toegevoegd die een literatuuroverzicht geven van de toxicologie, de farmacologie en de farmacotherapie van de actieve stof. De deskundigen die de rapporten opstellen kunnen zowel bij het bedrijf werkzaam zijn of van buiten komen.
Deel II - chemische, farmaceutische en biologische documentatie: eigenschappen, productie en houdbaarheid van de actieve stof en gerede product, bioequivalentie.
In deel II wordt voor de actieve stof met name de syntheseroute met de mogelijke bijproducten beschreven. Daarnaast worden de keuze van de hulpstoffen, het productieproces en het houdbaarheidsonderzoek met de resultaten daarvan beschreven. Alle specificaties zijn hier te vinden. Tevens wordt het bioequivalentie-onderzoek beschreven inclusief een uitvoerige statistische analyse.
Deel IV - Klinische documentatie: voor zover noodzakelijk.
Deel V - Bijzonderheden: inhoudsopgave, doseringsvormen, etikettering en productie- en marketingautorisatie. Voor generieke geneesmiddelen is het in het algemeen niet noodzakelijk om extra onderzoeken naar de toxicologie, de farmacologie en de farmacotherapie te overleggen. Deze gegevens staan in Deel III - Farmaco-toxicolologische documentatie, afkomstig uit de (medische) literatuur. Als alle documenten gereed zijn start het registratieproces met de indiening van het dossier bij het CBG, het College ter Beoordeling van Geneesmiddelen.
Fase 5: De registratieprocedure
Als alle documenten gereed zijn, kan het registratiedossier van het product ter beoordeling en goedkeuring worden ingediend bij het CBG. Deze beoordeelt alle gegevens op de aspecten werkzaamheid, veiligheid en kwaliteit.
In Europa bestaan er drie systemen om een geneesmiddel te registreren. Voor nieuwe actieve stoffen en biotechnologische (zogenaamde ‘high tech’) producten staat de centrale procedure via het Europese Agentschap EMEA (European Medicines Evaluation Agency) open. De EMEA is gevestigd in Londen. Voor bekende actieve stoffen, zoals generieke producten, wordt de decentrale procedure, ook wel "Wederzijdse Erkenningprocedure" of MRP (Mutual Recognition Procedure) gevolgd. Een derde mogelijkheid is de Nationale Procedure. Deze kan worden gevolgd als een geneesmiddel alleen in één land van de Europese Unie verkocht zal gaan worden en geen ‘high tech’ product is.
In de praktijk worden de meeste generieke producten geregistreerd via de MRP-procedure. Deze procedure houdt in dat het registratiedossier wordt aangeboden aan de autoriteiten van één van de lidstaten van de EU. Zij beoordelen het dossier en vragen waar nodig aanvullende informatie. Als zij vinden dat het dossier tezamen met de aanvullende informatie aan de kwaliteitsnormen voldoet, geven zij voor het product een handelsvergunning. Deze is alleen in het eigen land geldig. Vervolgens wordt een beoordelingsrapport opgesteld dat de fabrikant kan gebruiken om het registratiedossier in de andere EU-lidstaten in te dienen. In die landen is men vervolgens verplicht om het beoordelingsrapport van het referentieland te accepteren; zij mogen dus niet meer het gehele dossier beoordelen. Na de goedgekeurde MRP, beoordelen zij alleen het nationale deel, zoals de etikettekst en de tekst van de bijsluiter in de eigen taal. Daarna wordt een vergunning afgegeven. Alleen in het geval dat zij vinden dat toelating van het geneesmiddel de volksgezondheid in gevaar zou brengen, mogen zij weigeren een vergunning af te geven. In dat geval kan de fabrikant via een ingewikkelde en kostbare beroepsprocedure bezwaar aantekenen. In die procedure spelen de fabrikant en de registratieautoriteiten van het land dat het beoordelingsrapport heeft opgesteld een belangrijke rol.
De wederzijdse erkenningprocedure heeft als voordeel dat een heleboel beoordelingswerk slechts éénmaal gedaan hoeft te worden. Dat spaart tijd, menskracht en geld. In de praktijk blijkt echter dat individuele lidstaten - ondanks de positieve beoordeling door hun collega’s in een ander EU land - het dossier toch nog geheel of gedeeltelijk zelf gaan beoordelen en met aanvullende vragen komen. De voordelen van een Europese harmonisatie worden daarmee tenietgedaan.
Na registratie
Na het verkrijgen van de handelsvergunning is de fabrikant nog niet klaar. Meestal loopt het houdbaarheidsonderzoek dan nog. Bovendien kunnen bij het gebruik, de productie en de analyse van het geneesmiddel nieuwe inzichten ontstaan die aanpassingen in het registratiedossier noodzakelijk maken. Veel werk op een registratieafdeling heeft dan ook te maken met het actueel houden van de dossiers.